Bioinformatics work notes
Navigating the Genome
sequencing
-
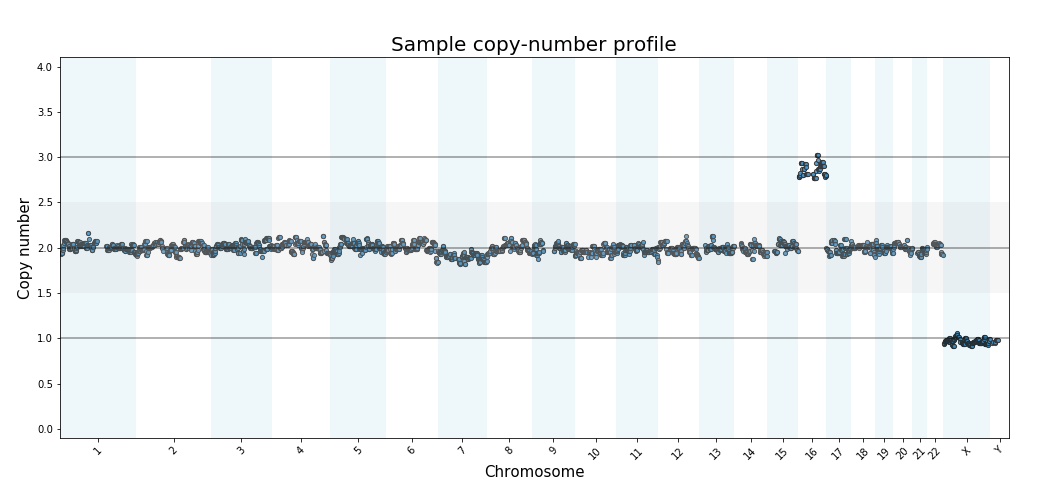
Read-counting for PGT-A
Read More: Read-counting for PGT-AIntroductory article about Pre-Implantation “Genetic Testing for Aneuploidy” using sequencing-based read-counting.
-
RNA-Seq data quality scores
Read More: RNA-Seq data quality scoresThere are different ways to encode the quality scores in FASTQ files from Next-generation…
-
Determining Coverage for NGS Data
Read More: Determining Coverage for NGS DataOne of the most common question after your Next-Generation Sequencing (NGS) run…
-
Command-line NGS data munging
Read More: Command-line NGS data mungingNotes about working with fasta / fastq files on the Unix command…
-
A DNA Sequencing History
Read More: A DNA Sequencing HistorySome of the landmarks in the history of DNA sequencing and molecular…
Gene-Test Bioinformatics Solutions GmbH
Expert advice, personalised service


